AKT,也称为磷酸激酶B (PKB),在涉及细胞生长和团结、细胞凋亡按捺和血管生成的多种级联信号传导机造中阐扬着关键感化。同时,AKT也显示出重要的代谢感化,此中包罗肌肉和脂肪细胞中的葡萄糖摄取或按捺神经元细胞灭亡等。
不外,关于处于PI3K/AKT/mTOR关键信号通路中心节点的AKT而言,因为PTEN缺失、AKT/PIK3CA突变或扩增等城市引起AKT信号通路的过度激活,招致肿瘤的发作和开展;近期研究更发现 AKT激活与肿瘤治疗中的耐药性相关。AKT由此成为肿瘤治疗的热门靶标。
AKT的发现和功用
AKT的名字由来也并不是基于它的功用,而是可能反映了AKT信号通路的发现过程。AKT最早能够文献逃溯至1977年,来自约翰霍普金斯大学肿瘤学中心的Stephen P. Staal博士在《美国国度科学院院刊》(PANS)上发文称,从自觉性淋巴瘤AKR/J小鼠的体外胸腺瘤细胞系AKT8别离到T-8病毒株。10年后,Stephen P. Staal博士再次颁发AKT8癌基因及其人类同源物的分子克隆,病毒基因组包罗病毒和非病毒的细胞相关序列,非病毒序列被定名为v-akt,揣测是AKT8病毒的病毒癌基因。因而,AKT 名字中的“Ak”是指发作自觉性胸腺淋巴瘤的AKR小鼠品系,“t”则代表“胸腺瘤”。那也招致后来确定了人源同基因/卵白时,AKT名字被继续沿用。
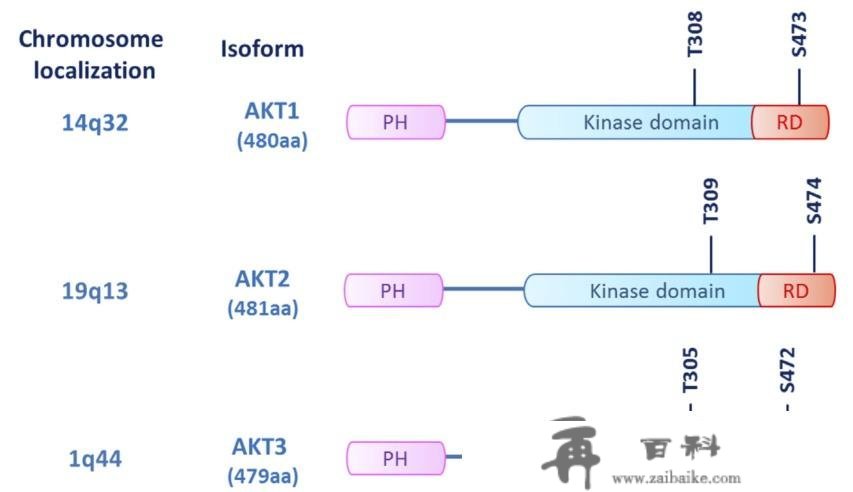
目前,AKT丝氨酸/苏氨酸激酶家族共发现了三种高度同源的亚型,即AKT1(PKBα)、AKT2(PKBβ)和 AKT3(PKBγ)。AKT三种亚型别离由39个氨基酸铰链区离隔的N端pleckstrin同源 (PH) 域和大的中央激酶域以及C端调理域 (RD) 构成。此中,PH域约有60%的同源性,而激酶构造域的同源性则超越85%。受多种细胞外刺激的刺激,各个AKT成员在差别情况中也具有普遍多样的下流效应。
Front. Pharmacol.12:662232
在散布表达上,AKT1和AKT2存在较为普遍。此中,AKT1通过按捺细胞凋亡过程参与细胞存活路子,能够阻遏细胞凋亡从而促进细胞存活,被认为是许多类型癌症的次要因素。AKT2的表达在褐色脂肪、骨骼肌和肝脏等胰岛素反响组织中升高,是胰岛素信号通路中的重要信号分子,诱导葡萄糖转运;AKT2缺失招致小鼠轻度生长缺陷并表示出糖尿病表型(胰岛素抵御)。AKT3似乎次要在大脑中表达,固然也在骨骼肌和肝脏中的表达,但程度较低,其感化不太清晰。在癌细胞中,AKT1 参与增殖和生长,促进肿瘤发作并按捺细胞凋亡;AKT2 调理细胞骨架动力学,有利于侵袭和转移;AKT3 过度激活在癌症中的感化仍然存在争议,虽然其存在刺激细胞增殖的可能。不外,在术语上PKB或AKT可能统称所有三个基因/亚型,但有时也用于零丁指代AKT1 / PKBα。
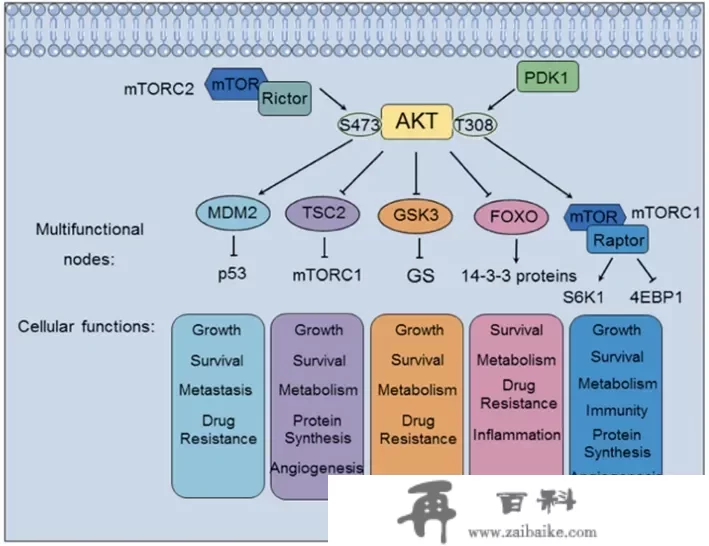
AKT1/2/3的次要磷酸化位点别离位于激酶域(T308/T309/T305) 和调理域 (S473/474/472) 中的苏氨酸和丝氨酸残基。AKT激活履历了双重磷酸化,也别离对应着上述激酶构造域(被PDK1磷酸化)和调理域(mTORC2磷酸化)。AKT激活后会磷酸化其下流靶标,包罗结节性硬化症复合体 2 (TSC2)、糖原合酶激酶-3β (GSK3β) 和叉头激酶转录因子 (FOXO),最末促进细胞增殖、代谢和存活。此外,AKT也能够基于ACK1或TNK2在其酪氨酸176残基处的磷酸化,招致其以不依赖PI3-激酶的体例激活。
Signal Transductionand Targeted Therapy (2021) 6:425
基于肿瘤基因测序的精准肿瘤学试验数据,AKT1-3突变在人类癌症突变中的占比约为3%-5%。此中,E17K是最常见、特征明白的激活突变,但在迄今为行的大型肿瘤测序研究中,E17K仅占AKT1-3突变的15%摆布。精准肿瘤学的目标,是将靶向治疗与驱动突变相婚配并制止在临床中利用突变抗性的药物,以进步患者的治疗反响和存活率。常见的E17K突变体和稀有的L52R、Q79K和D323H突变体都清晰地激活并驱动癌症转化。然而,与野生型AKT比拟,那些突变体都不会使细胞对变构或ATP合作性按捺剂更敏感。
靶向AKT:耐药治疗新战略
AKT相关的根底研究有比力高的学术起点,最早的文献记录是颁发在顶级期刊《美国国度科学院院刊》(PANS)上,原型AKT基因于1991年颁发在Science杂志上。也是因为一起头奠基的学术研究根底,AKT的转化开发引得浩瀚造药巨头喜爱。
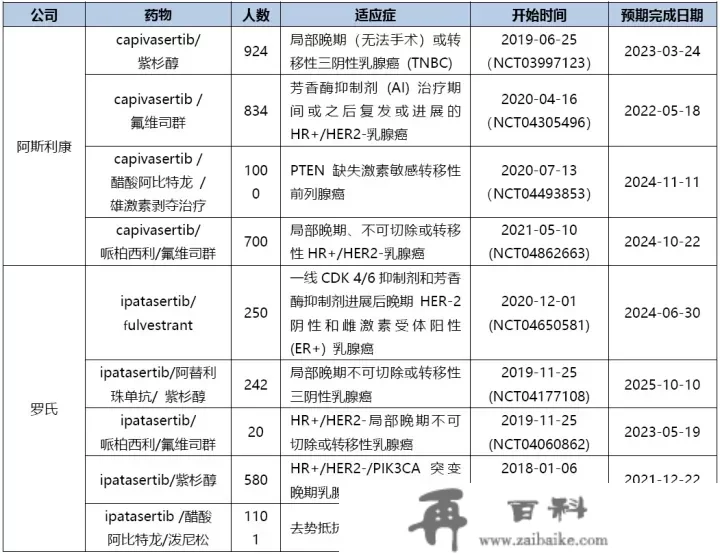
纵览AKT药物的开发过程,最早介入AKT按捺剂摸索和临床开发的公司是葛兰素史克、默沙东、礼来、拜耳,阿斯利康、罗氏(基因泰克)等一寡巨头。不外固然系出名门,但是那些公司开发的AKT按捺剂或者出卖,或者研发末行,如礼来的LY2780301、拜耳的BAY1125976等,在必然水平上也申明了新药研发的不确定性以及AKT信号通路的庞大复杂性和挑战性。默沙东的MK-2206,在停止了40多项早期试验后也最末折戟沉沙。
不断以来,内在或获得性耐药性是癌症药物疗效无法持续的次要原因,并最末招致治疗失败。因而,跟着对潜在机造的不竭摸索,发现更有前景的药物靶点正成为癌症治疗和预防的更佳治疗办法。PI3K/AKT通路的异常激活以及上游或下流靶标转导,做为负责多种肿瘤构成耐药性的重要信号通路起着至关重要的感化。
为了加强肿瘤细胞的敏感性和匹敌肿瘤的耐药性,AKT按捺剂在临床研究中也获得必然停顿。例如,在乳腺癌中,PI3K/AKT 通路被认为是内排泄治疗耐药性开展的关键参与者,联用AKT按捺剂匹敌PI3K/AKT通路的激活,进而通过阐扬细胞毒活性来阻遏肿瘤细胞的耐药性。因而,阿斯利康、罗氏也在评估零丁或组合利用AKT按捺剂,以开发肿瘤治疗新疗法以及应对治疗抗性。
来源:医药魔方NextPharma数据库
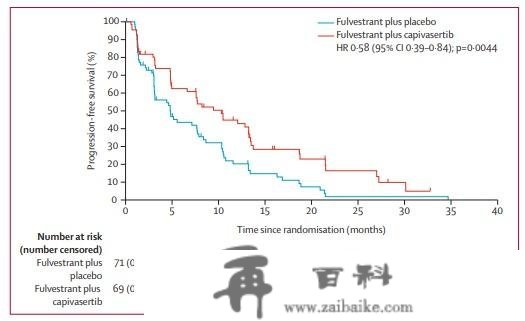
ipatasertib和capivasertib是全球最早进入临床III期开发的AKT按捺剂,适应症次要集中在HR+乳腺癌、三阴性乳腺癌和前列腺癌。2020年,阿斯利康披露了capivasertib结合氟维司群治疗芳香酶按捺剂治疗后复发或停顿的HR+/HER2-乳腺癌临床II期数据(FAKTION)。研究成果显示,随机承受氟维司群加capivasertib (n=69) 治疗的患者相较于或氟维司群加慰藉剂 (n=71) ,mPFS显著耽误(10.3 vs 4.8 m),降低疾病停顿风险42%(HR:0·58,95%CI 0.39–0.84),到达研究的次要起点。
来凯医药:biotech中的AKT按捺剂开发先行者
capivasertib的核心关键注册性临床研究将于明年完毕,或许AKT按捺剂的“破茧成蝶”、药物上市的拂晓时刻即将到来。在全球biotech企业中,来凯医药在AKT按捺剂afuresertib临床停顿最快,已经处于关键的II期注册研究,具有必然的先发优势。
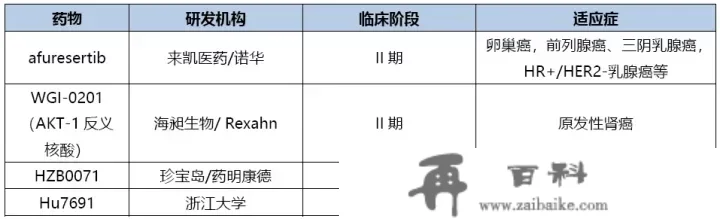
来源:医药魔方NextPharma数据库
来凯医药优先规划AKT按捺剂与其开创人吕朝阳博士的履历不无关系。吕朝阳博士结业于南开大学,在美国北卡罗来纳大学教堂山分校获得生物化学博士学位,并在美国哈佛大学完成博士后,是最早参加诺华生物医学研究中心(Novartis Institutes for BioMedical Research, NIBR)的科学家之一。他在诺华工做过13年,期间率领研发团队完成了二十余项新药研发项目。
诺华的两款AKT按捺剂afuresertib和uprosertib最早均由GSK开发,先后完成了卵巢癌、胃癌、多发性骨髓瘤、黑色素瘤等适应症范畴的20多项临床I/II期研究,药物的临床疗效和耐受平安性得到初步临床验证。因为GSK与诺华在2014年达成了资产置换协议,Afuresertib和uprosertib随之归诺华所有。不外,诺华其时在肿瘤范畴正专注于“变化性”的CAR-T细胞疗法和基因疗法,AKT按捺剂在继续做了4年临床试验之后,犹如K药被收买后的命运一般,被“束之高阁”。
其实,诺华研发部分有良多科学家已经看到了AKT激酶选择性按捺剂的潜在价值,此中就包罗吕朝阳博士。当诺华起头启动对外受权时,来凯医药占据了天时人地相宜,先后拿到了afuresertib(LAE002)和uprosertib(LAE003)的权益。尤其值得一提的是,来凯医药拿到的是那两个分子的全球权益,而非部门区域。来凯也由此成为biotech中少有的重点规划AKT按捺剂的公司,属于那个范畴的先行者。
随之而来的却是适应症选择问题,AKT按捺剂事实可以处理哪些范畴的未满足需求?

历来凯医药的产物管线中,我们不难看出,afuresertib(LAE002)选择卵巢癌优先开发。其实,那不只表现了适应症上的差别化,也确实是契合AKT按捺剂感化机造和临床需求的未满足。在差别瘤种中,卵巢癌都是AKT1-3过表达的优势瘤种。
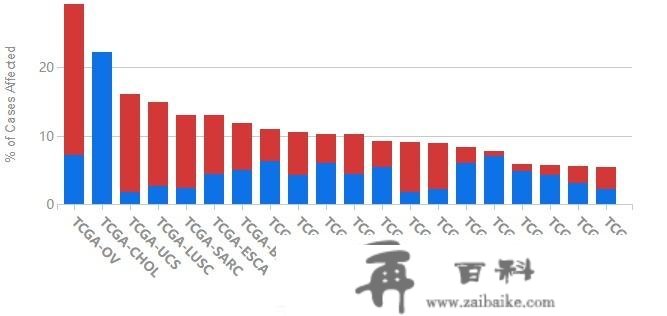
OV:卵巢浆液性囊腺癌(来源:TCGA)
浆液性囊腺癌是卵巢恶性肿瘤中最常见的类型。铂类结合紫杉醇类药物当前仍是卵巢癌治疗的首选药物,不外大约10%-15% 的卵巢癌患者对铂化疗无反响;有效患者中,也有80%以上会复发,最末会呈现耐药性。上述疗法耐药之后,仅靠非铂类化疗药物治疗,有效性难以令人满意。当然,贝伐珠单抗结合紫杉醇的胜利,以及PARP按捺剂临床研究,使结合用药战略成为进步药物敏感性和匹敌耐药的潜力战略。然而,贝伐珠单抗+化疗目前仅限于颠末不超越2线治疗的患者,在美国,其现实利用仅限于部门的卵巢癌患者,后期的治疗仍然回到化疗。
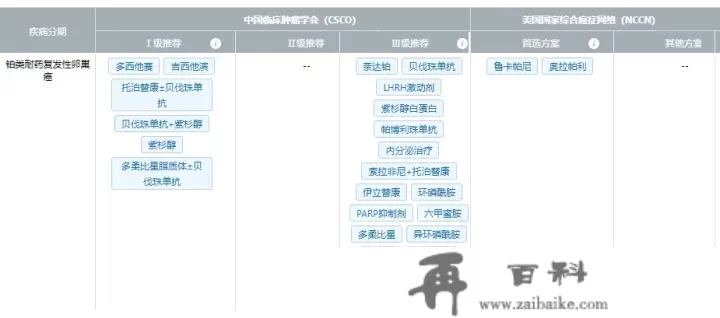
来源:医药魔方DeepMed数据库
PI3K/AKT通路的异常激活在多种肿瘤构成耐药性的重要信号通路中起着至关重要的感化,因而,afuresertib选择铂类耐药复发性卵巢癌也算是“理所应当”。临床前的研究显示卵巢癌患者在铂和紫衫醇的治疗中,经常会因为AKT的过火激活而招致耐药,此时按捺AKT能够从头恢复患者对化疗的敏感性。早期的临床试验也令人信服的验证了那一耐药机理,在多线铂治疗耐药的卵巢癌患者中,afuresertib结合卡铂和紫衫醇显示了临床显著的抗肿瘤疗效和优良的平安性。
当然,在前列腺癌、三阴乳腺癌,HR+/HER2-乳腺癌范畴,afuresertib也并未放弃。针对HR+/HER2-乳腺癌,afuresertib已经在中美两国,获批了一项Ib/III期全球多中心临床研究;抗PD-1/PD-L1耐药的特定实体瘤、三阴乳腺癌也已经推进至I/II期研究。
对准PD-1/PD-L1耐药
2021年7月,来凯医药颁布发表与信达生物达成合做协议,配合开展afuresertib与信迪利单抗结合用药的临床研究2022年1月,两边合做的治疗抗PD-1/PD-L1耐药特定实体瘤患者的结合治疗计划临床I/II期试验申报,获得了中国国度药品监视办理局药品审评中心(CDE)的批准。
此次临床研究对准的是当前大热的PD-1/PD-L1肿瘤免疫疗法的耐药性问题。在来凯医药首席医学官岳勇博士看来:虽然具有持久的、潜在的接近于治愈的临床好处,但耐药性仍然是障碍PD-1/PD-L1疗法进一步应用的严重挑战。因而,免疫抵御是一种未竟的庞大医疗需求,是进步癌症患者保存率和生活量量的次要障碍之一,而癌细胞中的AKT激活是可能招致免疫抵御的浩瀚因素之一。
结合计划接纳三个药物:来凯医药的泛AKT按捺剂——处于临床开发阶段的1类候选新药afuresertib(LAE002)、信达生物的信迪利单抗打针液以及化疗药物白卵白连系型紫杉醇或多西他赛。来凯医药的泛AKT按捺剂具有潜在的平安性优势,相较于其他临床在研产物的间歇给药计划,afuresertib能够实现口服每日给药。也恰是那种平安性优势,使afuresertib更具备结合应用潜力。
该项研究的目的旨在I期部门评估上述三种药物联用的平安性、确定II期研究保举剂量,并在 II 期研究中评估结合治疗的临床有效性和平安性。II 期的研究人群为对既往抗 PD-1/PD-L1 治疗(单药治疗或与其他抗癌药物结合治疗)耐药且患有以下5种选定肿瘤之一的患者:
非小细胞肺癌(NSCLC)胃和食管胃连系部腺癌(GC/GEJC)食道癌(EsC)宫颈癌(CC)子宫内膜癌(EC)结语
纵不雅AKT按捺剂的研究历程,不乏高低和坎坷。曾经浩瀚跨国巨头的相继进入,似乎也令AKT“风光无二”;先后的折戟沉沙,使AKT按捺剂开发者事与愿违。得益于阿斯利康、罗氏和来凯医药的对峙与开辟,AKT按捺剂将在2022年再次迎来关键时刻,能否成为“破茧成蝶”的光亮时刻,让我们拭目以待。
注:原文有删减
参考材料
[1] What Does the“AKT” Stand for in the Name“AKTKinase”? Some Historical Comments. Comments. Front. Oncol. 10:1329
[2] Staal SP, et al. Proc Natl Acad Sci USA.(1977) 74:3065–7; Proc Natl Acad Sci USA. (1987) 84:5034–7
[3] AKT Inhibitors: New Weapons in the FightAgainst Breast Cancer? Front. Pharmacol. 12:662232
[4] AKT/PKB Signaling: Navigating the Network. Cell 169, 2017: 381
[5] Recurrent AKT mutations in human cancers: functional consequences and effects on drug sensitivity. Oncotarget. 2016 Jan26; 7(4): 4241–4251
[6] A Portrait of AKT Kinases. Cancer Biology & Therapy 3:3, 268-275
[7] AKT inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). INTERNATIONALJOURNAL OF ONCOLOGY 48: 869-885; The AKT pathway in oncology therapy and beyond.INTERNATIONAL JOURNAL OF ONCOLOGY 53: 2319-2331
[8] The AKT/PKB pathway: molecular targetfor cancer drug discovery. Oncogene (2005) 24, 7482–7492
[9] AKT/Protein Kinase B Isoforms Are Differentially Regulated by Epidermal Growth Factor Stimulation. THE JOURNAL OF BIOLOGICAL CHEMISTRY, 40: 30934–30942
[10] A. Bellacosa, J.R. Testa, S.P. Staal,P.N. Tsichlis, A retroviral oncogene, AKT, encoding a serine-threonine kinase containing an Sh2-like region. Science 254(1991) 274–277
[11] Phase 1 Dose Escalation Study of the Allosteric AKT Inhibitor BAY1125976 in Advanced Solid Cancer-Lack of Association between Activating AKT Mutation and AKT Inhibition-Derived Efficacy. Cancers (Basel). 2019 Dec10;11(12):1987
[12] A first-in-human phase I trial of LY2780301, a dual p70 S6 kinase and Akt Inhibitor, in patients with advanced or metastatic cancer. Invest New Drugs. 2015 Jun;33(3):710-9; Safety, tolerability and antitumour activity of LY2780301 (p70S6K/AKT inhibitor) in combination with gemcitabine in molecularly selected patients with advanced or metastatic cancer: a phase IB dose escalation study. Eur J Cancer . 2017 Sep;83:194-202
[13] The Pathogenic Role of PI3K/AKT Pathwayin Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949
[14] Fulvestrant plus capivasertib versusplacebo after relapse or progression on an aromatase inhibitor inmetastatic,oestrogen receptor-positive breast cancer (FAKTION):a multicentre,randomised, controlled, phase 2 trial. Lancet Oncol 2020; 21: 345–57
[15] Targeting PI3K/Akt signal transductionfor cancer therapy. Signal Transduction and Targeted Therapy (2021) 6:425
[16] Innovative therapies to tackleplatinum-resistant ovarian cancer. Nature 600, S45-S47 (2021)
免责声明:本文仅做信息交换,文中的概念不代表中肽生化立场,亦不代表中肽生化撑持或反对文中概念。本文也不是研究治疗计划保举。若是需要获得治疗计划指点,请前去正规病院就诊。
来源:医药魔方